多肌炎和皮肌炎的基础知识
导读:病人将上肢举过肩部,上台阶及从坐位站起时感到困难,由于骨盆带和肩带肌群无力,病人需要坐轮椅或卧床不起。

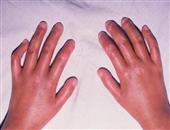
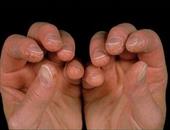
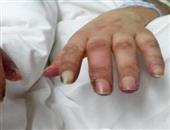
多肌炎为系统性结缔组织疾病,特点是肌组织出现炎症,变性等改变(皮肤也常同样受累、即皮肌炎),结果导致对称性肌无力和一定程度的肌萎缩,主要在肢带肌。
肌炎分类包括原发性特发性多肌炎。儿童型皮肌炎或多肌炎。成人型原发性特发性皮肌炎。包涵体肌炎。合并恶性肿瘤的皮肌炎或多肌炎。多肌炎或皮肌炎合并各种结缔组织病的重叠综合征,包括混合性结缔组织病和硬化性皮肌炎。多肌炎和皮肌炎的某些临床表现与进行性系统性硬化症相同,有时与sle或血管炎相同。
发病可以呈急性或隐匿起病。急性感染可为其前驱表现或发病的诱因。成人与儿童症状类似,但儿童发病往往很急,而成人多为隐匿发病。早期症状通常为近端肌无力或皮疹。(在包涵体肌炎,远端肌也累及甚至比近端肌更重)。肌肉触痛和疼痛明显少于肌无力。皮疹,多关节痛,雷诺现象,吞咽困难,肺部疾病及全身不适,发热,乏力,体重下降等均可出现。
肌无力可突然发生,并持续进展数周到数月以上。它能破坏50%肌纤维,从而引起肌无力(肌无力说明进展性肌炎)。病人将上肢举过肩部,上台阶及从坐位站起时感到困难,由于骨盆带和肩带肌群无力,病人需要坐轮椅或卧床不起。颈屈肌可严重受累,不能从枕头上将头抬起。喉部肌肉无力是造成发音困难的原因。胸壁肌肉和膈肌的受累可引起急性呼吸功能不全。咽、食管上端横纹肌受累引起吞咽困难和反流。食管下段和小肠蠕动减弱与扩张,同进行性系统性硬化症所见难以区别。手、足、面部等肌肉一般不受累。肢体挛缩可发生在疾病后期的慢性阶段。
点赞(0)
分享到: